
У человека ген вызывающий потемнение зубов доминантен и локализован
Опубликовано: 04.05.2024

Весь контент iLive проверяется медицинскими экспертами, чтобы обеспечить максимально возможную точность и соответствие фактам.
У нас есть строгие правила по выбору источников информации и мы ссылаемся только на авторитетные сайты, академические исследовательские институты и, по возможности, доказанные медицинские исследования. Обратите внимание, что цифры в скобках ([1], [2] и т. д.) являются интерактивными ссылками на такие исследования.
Если вы считаете, что какой-либо из наших материалов является неточным, устаревшим или иным образом сомнительным, выберите его и нажмите Ctrl + Enter.
- Код по МКБ-10
- Эпидемиология
- Причины
- Факторы риска
- Патогенез
- Симптомы
- Формы
- Осложнения и последствия
- Диагностика
- Дифференциальная диагностика
- Лечение
- К кому обратиться?
- Профилактика
- Прогноз

Относительно редкое заболевание – эктодермальная дисплазия – являет собой генетическое нарушение, сопровождающееся расстройством функциональности и структуры производных элементов наружного слоя кожи. Обычно поражаются волосы, ногтевые пластины, зубы, железистая система (слизистая, потовая и сальная). Болезнь сложная, может протекать в нескольких формах. Лечение преимущественно корректирующее, симптоматическое: к сожалению, о полном выздоровлении речь не идет. [1]
Код по МКБ-10
Эпидемиология
Вариациями эктодермальной дисплазии являются синдром Криста-Сименса-Турена, а также синдромы Клоустона, Рэппа-Ходжкина и ЕЕС. Патология впервые была описана в середине XIX века доктором Туреном. В 1913 и 1929 годах это описание было дополнено соответственно врачом-стоматологом Кристом и дерматологом Сименсом, в 1968 году – Рэппом и Ходжкиным, в 1970 году – Рудигером.
В медицинской литературе заболевание чаще встречается именно под названием эктодермальной дисплазии и соответствует международной кодировке Q82.4 (МКБ-10).
На сегодняшний день точную частоту заболеваемости врачи озвучить не могут. Однако считается, что синдром встречается примерно в одном случае из 5-10 тысяч. [2]
Точно известна этиологическая гетерогенность заболевания с тремя генетическими типами наследственной передачи: аутосомно-рецессивный, доминантный и Х-сцепленный рецессивный (последний – наиболее распространенный). [3]
В данный момент определены три гена в различных хромосомах, позволяющих выявить данное заболевание при помощи генетико-молекулярных методик. Численность возможных мутаций – более шестидесяти.
Эктодермальной дисплазией болеют чаще мальчики, что связано со сцепленным с полом наследованием. Девочки зачастую имеют легкую форму патологии, либо бессимптомную.
Патология регистрируется в разных странах мира у представителей любой расы. Может спорадически появляться у клинически здоровых пар, либо проявляться в семейной форме (особенно часто – если родители имеют близкородственные связи).
Причины эктодермальной дисплазии
Единственная причина развития эктодермальной дисплазии заключена в мутации определенного наследственного генного фактора. В частности, наиболее распространено нарушение со стороны гена EDA, локализованного на Х-хромосоме. Этот ген ответственен за кодировку белкового вещества эктодисплазин-А, неправильная структура которого влечет за собой нарушение формирования элементов эктодермы. В настоящее время точные характеристики белкового вещества и механизм развития мутационных нарушений не выяснены.
Заболевание, сцепленное с Х-хромосомой, имеет свои особенности: проблема обнаруживается чаще у мужчин, но и женщины способны выступать не просто носителями, но и иметь отдельные признаки синдрома, хоть и в нетяжелой степени. К примеру, у пациенток с эктодермальной дисплазией можно наблюдать избыточную сухость кожных покровов, морщинистость, истончение и сухость волос, зубную деформацию. Возможны проблемы с грудными железами и сосками. Подобные признаки указывают на возможность неполного доминирования генных мутаций EDA.
Среди других типов мутаций можно выделить изменения гена EDAR, который отвечает за кодировку рецептора к фактору опухолевого некроза. Этот ген локализуется на II хромосоме, наследование происходит аутосомно-рецессивно. В точности процесс развития патологии не выяснен.
Если речь идет о редких вариантах эктодермальной дисплазии, то они возникают под влиянием генных мутаций в TDARADD, ответственном за кодировку белка-рецептора к экзодисплазину-А, локализованному на I хромосоме. Патогенетические механизмы полностью не изучены. [4]
Факторы риска
Наиболее значимыми факторами риска, ведущими к рождению ребенка с эктодермальной дисплазией, являются дефекты:
- гена EDA, кодирующего эктодисплазин-А, картированного на хромосоме Xq12-q13.1;
- гена EDAR, кодирующего рецептор фактора опухолевого некроза, члена суперсемейства EDAR, картированного на хромосоме 2q11-q13;
- гена TDARADD, кодирующего эктодисплазин-А – рецептор-ассоциированный белок, картированного на хромосоме 1q42.2-q43.
Заподозрить наследственную предрасположенность к эктодермальной дисплазии можно при исследовании семейной истории.
Комплексная генно-молекулярная диагностика позволяет оценить генетический риск развития у ребенка данного синдрома.
Патогенез
Патогенетические особенности развития данного заболевания малоизученны. Известно, что эктодермальная дисплазия появляется в результате мутационных изменений в определенных генах. Причина наиболее частой формы патологии заключается в повреждении гена EDA, локализованного на Х-хромосоме. Этот ген отвечает за кодировку белкового агента с названием эктодисплазин-А. Патологические изменения в его структуре вызывают неправильное развитие эктодермальных производных. К сожалению, на сегодняшний день и функциональная сторона этого белкового агента, и патогенез изменений при мутировании гена EDA изучены недостаточно.
Основная отличительная черта эктодермальной дисплазии заключается в том, что клинические нарушения обнаруживаются не только у пациентов мужского пола, но и у женщин: носительство проявляется более легкими патологическими изменениями. В частности, отмечается сухость волос и кожных покровов, ранняя морщинистость, искривления и прочие нарушения со стороны зубов.
Помимо этого, причинами типичного синдрома Криста-Сименса-Турена выступают мутационные изменения в гене EDAR, ответственном за кодировку одного из рецепторов к фактору опухолевого некроза. Этот ген локализован на II хромосоме, наследование происходит аутосомно-рецессивно. Патогенетические особенности и в этом случае не изучены. [5]
Известна и более редкая разновидность ангидротического типа эктодермальной дисплазии, с аутосомно-доминантным способом наследования. Причиной выступают мутационные изменения гена TDARADD, кодирующего белковое вещество-рецептор к экзодисплазину-А и локализованного на I хромосоме. Скорее всего, патогенетические характеристики в данном случае идентичны с более распространенной разновидностью заболевания, сцепленной с полом.
К сведению: эктодерма представляет собой один из трех зародышевых листков (ещё два представлены мезодермой и энтодермой). Эктодерма – это наружный слой, который формируется на протяжении третьей недели эмбрионального развития и обусловливает образование кожных покровов и придатков (волосы, ногти), прямокишечного и ротового эпителия, зубной эмали, хрусталика и роговицы, потовыделительных желез. У людей, страдающих эктодермальной дисплазией, некоторые или все структуры эктодермы, либо отсутствуют, либо недостаточно функционируют.
Симптомы эктодермальной дисплазии
Клиническая картина при эктодермальной дисплазии определена целым рядом многочисленных нарушений, затрагивающих эктодерму и потовые железы. Поражаются также сальные и апокриновые железы, но эти дефекты менее выражены. Другие железистые системы – в частности, слезные, пищеварительные, носовые, бронхиальные – имеют признаки атрофии. Типичные признаки: атрофическое процессы, гипоплазия кожных покровов, гипоплазия грудных желез и сосков.
В области лица обнаруживаются морщины, истончение век, периорбитальное нарушение пигментации, папулы, экзематозные высыпания, ладонный гиперкератоз. Лобные бугры и надбровные дуги явно выдаются вперед, переносица сглажена, нос небольшой седловидный, носовые крылья гипопластичны, губы полные и выступающие, щеки – впалые.
Волосяной покров редкий, часто – с залысинами, отличается сухостью и светлыми оттенками.
Зубы имеют неправильную конфигурацию, часто – коническую заостренную форму. Некоторые зубы вовсе отсутствуют (клыки присутствуют всегда).
Ушные раковины также деформированы: обычно они маленькие, высоко посаженные, ушной завиток имеет неправильную форму.
Со стороны органов зрения может отмечаться помутнение хрусталика, близорукость, блефароконъюнктивит, снижение слезовыделения, жидкое стекловидное тело.
Некоторые пациенты страдают полным отсутствием слуха. Отмечается склонность к инфекционным заболеваниям, нарушениям терморегуляции.
Первые признаки
Первые проявления эктодермальной дисплазии зачастую обнаруживаются уже в периоде новорожденности. Однако это может произойти и позже, так как клиническая картина у маленьких детей не всегда носит выраженный характер и усугубляется с годами.
Базовыми симптомами, по которым можно заподозрить наличие патологии, часто становятся такие:
- отсталость роста на фоне относительно большой головы;
- сухость, тонкость волос, преимущественно «пушковые» волосы с замедленным ростом и малой пигментацией, укороченность и редкость ресниц и бровей, либо полное их отсутствие;
- ранняя алопеция, вплоть до полной потери волосяного покрова;
- типичный внешний вид по типу «стариковского лица», выступающая лобная область, надбровные дуги и бугры, расширенные скулы, западение переносицы, небольшой седлообразный нос и гипопластические крылья, впалые щеки, выдающиеся губы по типу «рыбьих», «тяжелый» подбородок, неправильная форма ушных раковин;
- запоздалое прорезывание зубов (от года до трех лет), нарушение привычной последовательности прорезывания, длительный период сохранения молочных зубов, отсутствие некоторых зубов;[6]
- конусовидная зубная конфигурация, заостренные режущие края, сглаженная поверхность моляров;
- нарушения зубного ряда и прикуса;
- недостаточно развитые слюнные железы, слабое слюновыделение, сухость во рту, сиплость;
- чрезмерная сухость кожи, ранняя морщинистость, что особенно заметно на лице;
- нарушения пигментации, неправильная работа сальных желез, папулезная сыпь;
- конъюнктивиты, светофобия;
- недоразвитые грудные железы, либо их отсутствие;
- недостаточно развитые слизистые железы в респираторной и пищеварительной системе, что обусловливает частые бронхиты, риниты, синуситы, желудочно-кишечные патологии;
- периодическое резкое повышение температуры, связанное с неправильной теплоотдачей из-за нарушения работы потовых желез;
- реже – умственная отсталость, олигофрения (чаще развитие интеллекта соответствует норме);
- нарушенная социальная адаптация и ориентация, скованность и замкнутость;
- речевые проблемы, связанные с неправильным зубным ростом и сухостью слизистых ротоносоглотки;
- нарушенное зрение;
- слабое или отсутствующее потовыделение.
Триада при ангидротической эктодермальной дисплазии
Ангидротический вариант эктодермальной дисплазии проявляется триадой базовых признаков:
- редкое оволосение по типу атрихоза или гипотрихоза; [7], [8]
- неправильная конфигурация зубов (часто – коническая, заостренная), либо недоразвитие и отсутствие зубов;
- нарушения потовыделения по типу гипо и ангидроза, что часто обусловлено отсутствием потовых желез как таковых.
Ввиду наличия ангидроза у больного отмечаются такие вспомогательные признаки, как высокотемпературная гиперчувствительность и регулярные рецидивы гипертермии, что представляет собой реальную опасность для жизни человека. Кожные покровы истончены, отличаются сухостью. Многие больные страдают хроническим блефароконъюнктивитом, «синдромом сухого глаза», астмоподобными состояниями. [9]
Формы
Разные совокупные проявления и их интенсивность определяют подразделение эктодермальной дисплазии на несколько видов, которые можно назвать самостоятельными формами патологии. Основными такими видами считаются: синдром Криста-Сименса-Турена, синдром Клоустона, синдром Рэппа-Ходжкина и синдром ЕЕС.
Синдром Криста-Сименса-Турена, или ангидротическая эктодермальная дисплазия характеризуется полной дисфункцией потовых желез, а также своеобразным фенотипом строения лицевой области: у ребенка обнаруживается выдающийся кпереди лоб, утонченные и негустые брови, редкие короткие ресницы, морщины. Типично периорбитальное нарушение пигментации, седлоподобная переносица, челюстная гипоплазия. Волосы бывают депигментированными, либо слабопигментированными.
Некоторые специалисты посчитали, что полномерный ангидроз у пациентов все же обнаруживается редко, и у большинства больных потовыделительная система слабо, но все-таки функционирует. Это мнение было учтено и привело к тому, что врачи со временем стали использовать более корректное название: гипогидротическая форма заболевания. Гипогидротическая эктодермальная дисплазия представляет собой генетическое расстройство формирования эктодермального слоя. Патология характеризуется нарушениями образования таких элементов эктодермы, как кожные и волосяные покровы, железы (потовые, сальные) и зубы. Болезнь состоит из трех подтипов, которые практически не отличаются симптоматически, так как основным клиническим признаком становится нарушенное потовыделение (преимущественно гипогидроз). Речь идет о непосредственно синдроме Криста-Сименса-Турена с Х-сцепленным типом наследования, а также аутосомно-рецессивной и аутосомно-доминантной эктодермальной дисплазии. Существует ещё несколько менее распространенных подтипов, сопровождающихся выраженным дефицитом иммунитета – так называемая врожденная ангидротическая эктодермальная дисплазия с иммунодефицитным состоянием.
Синдром Клоустона представляет собой гидротический тип эктодермальной дисплазии. Определяющими симптомами патологии становятся все те же поражения зубов, волос и потовыделительной системы, но в несколько меньшей степени. Гиподентия обнаруживается в области нижних резцов, вторых моляров и верхних клыков. Ногтевые поражения проявляются в виде гипоплазии, дистрофии, аплазии с паронихиями. Снижена численность потовыделительных желез, при неизменных сальных железах. Возможен гипотрихоз, облысение. Способ наследования аутосомный и аутосомно-доминантный.
Синдром Рэппа-Ходжкина иначе называют гипогидротической эктодермальной дисплазией, сопровождающейся расщелиной губы, альвеолярного отростка, мягкого и твердого неба. Отличительными проявлениями считаются такие: гипогидроз и гипотрихоз, патологические изменения ногтей, гиподентия или олигодентия в соединении с расщелиной верхней губы, альвеолярного отростка, мягкого и твердого неба. Общими симптомами становятся также впадение переносицы, сужение носа, микрогнатия верхней челюсти, небольшой рот, уменьшенные гениталии. Синдром наследуется по аутосомно-доминантному типу.
Синдром ЕЕС лишь недавно выделили в качестве самостоятельного заболевания, более известного как совокупный синдром эктродактилии, эктодермальной дисплазии с расщелиной неба и верхней губы. Отличительными симптомами являются дефекты стоп и кистей, расщелина верхней губы, иногда – расщелина языка. Эти признаки присутствуют на фоне нарушения потовыделения, гипотрихоза и алопеции, ногтевой гипоплазии, сухости и гипопигментации кожи, конъюнктивита, светофобии и пр. Типичны также зубные аномалии, позднее прорезывание, множественный кариес. На фоне физических дефектов умственное развитие обычно адекватное. Способ наследования – аутосомно-доминантный, но встречаются и рецессивно наследованные варианты.
Эктодермальная дисплазия у детей
Несмотря на то, что эктодермальная дисплазия является врожденным заболеванием, её не всегда удается диагностировать у новорожденного малыша: зачастую диагноз ставят лишь через несколько лет (часто – к 2-3 годам). Практикующие врачи отмечают необходимость ранней диагностики, так как от этого может зависеть не только дальнейший образ жизни, но иногда и непосредственно жизнь больного. Симптомы патологии многообразны, но не всегда заметны. При этом некоторые из них встречаются чаще, а другие – реже. [10] И родителей, и врачей должны насторожить такие признаки:
- гипоплазия потовыделительных желез с гипо или ангидрозом, терморегуляторные нарушения, частые эпизоды гипертермии, беспричинная лихорадка, регулярные перегревы;
- явления гипотрихоза, редкость, депигментация и тонкость волос, укорочение бровей и ресниц (или их отсутствие);
- устойчивое или преходящее облысение (полное или очаговое);
- позднее зубное прорезывание с нарушением его последовательности;
- недостаток количества зубов, нарушение их конфигурации (часто – конусообразная, шиповидная форма с заостренным краем), либо отсутствие зубов;
- нарушения прикуса, иногда – подвижность зубов, большие межзубные промежутки;
- низкое прикрепление верхнегубной уздечки, резкая выраженность щечных тяжей, мелкое ротовое преддверие;
- недостаточно развитый верхнечелюстной альвеолярный отросток;
- на рентгенограмме – укороченные зубные корни, расширенные периодонтальные щели, уплощенные мыщелковые нижнечелюстные отростки;
- гипоплазия слизистых желез во рту, как результат – недостаточное слюновыделение, сиплость;
- склонность к грибковым стоматитам, хейлитам;
- типичное «лицо старика» с выдающейся лобной областью, запавшей переносицей, небольшим седлообразным носом, впалыми щеками, полными нечеткими выпуклыми губами, неправильной формой ушных раковин;
- истонченная сухая морщинистая кожа, иногда – с папулезной сыпью;
- недостаточная функция слезных желез, частые воспалительные заболевания (кератиты, блефариты и пр.);
- дефекты губы и неба;
- ногтевые поражения, паронихии;
- дефекты стоп и/или кистей, гиперкератоз ладоней и стоп;
- недостаточное развитие грудных желез и сосков (иногда – их отсутствие);
- иммунодефицит, экзема;
- склонность к респираторным и пищеварительным заболеваниям, а также к носовым кровотечениям.
Разные сочетания признаков и их интенсивность определяют отдельные варианты течения эктодермальной дисплазии.
Классическая гемофилия передается как рецессивный, сцепленный с Х -хромосомой, признак. Мужчина, больной гемофилией, женился на здоровой женщине (все ее предки были здоровы). У них родилась здоровая дочь. Определить вероятность рождения больного гемофилией ребенка от брака этой дочери со здоровым мужчиной.
А – нормальная свертываемость, а – гемофилия.
- Мужчина болен гемофилией, следовательно, его генотип – Х а Y .
- Женщина здорова, значит, она несет доминантный ген А . Все ее предки были здоровы (чистая линия), следовательно, она не является носительницей, и ее генотип – Х A Х A .
- Одну Х -хромосому дочь получила от матери, другую от отца. Мать могла передать ей только хромосому Х A , а отец – только Х a . Генотип дочери – Х A Х a .
- Генотип мужа дочери – Х A Y , по условию задачи.
Схема брака
| P | ♀ Х A Х a здорова | × | ♂ Х a Y гемофилия | ||
| гаметы |  |   | |||
| F1 | Х A Х a носитель | Х A Y здоров | |||
| гаметы | | | | | |
| F2 | Х A Х A здорова 25% | Х A Х a носитель 25% | Х A Y здоров 25% | Х a Y гемофилия 25% | |
Вероятность рождения больного гемофилией ребенка – 25% (50% мальчиков будут страдать этим заболеванием).
У дрозофилы доминантный ген красной окраски глаз ( W ) и рецессивный ген белой окраски ( w ) находятся в Х -хромосомах. Белоглазая самка скрещивалась с красноглазым самцом. Какой цвет глаз будет у самцов и самок в первом и втором поколении?
Отсутствие потовых желез у людей – рецессивный признак, сцепленный с Х -хромосомой. Мужчина, у которого отсутствуют потовые железы, женился на женщине, в семье которой никогда не встречалось это заболевание. Какова вероятность рождения у них детей с подобной аномалией?
У человека гемофилия детерминирована сцепленным с Х -хромосомой рецессивным геном. Какова вероятность рождения больного ребенка от брака с генотипически здоровым партнером:
а) мужчины, брат которого страдает гемофилией;
б) здоровой женщины, имеющей такого брата?
Рецессивный ген дальтонизма (цветовой слепоты) располагается в Х -хромосоме. Женщина с нормальным зрением (отец ее был дальтоником) выходит замуж за мужчину с нормальным зрением, отец которого был дальтоником. Определить возможные фенотипы потомства.
Рецессивный ген дальтонизма локализован в Х -хромосоме. От брака женщины с нормальным зрением, родственники которой страдали дальтонизмом, и мужчины с нормальным зрением, у отца которого была цветовая слепота, родились три дочери с нормальным зрением и два сына с цветовой слепотой. Каковы генотипы родителей и потомства? От кого из родителей мальчики получили ген дальтонизма?
У человека цветовая слепота обусловлена рецессивным геном, сцепленным с Х -хромосомой. Нормальное зрение определяется доминантным аллелем этого гена. От брака родителей с нормальным зрением родился ребенок с цветовой слепотой. Определить генотипы всех членов семьи.
У дрозофилы есть пара аллельных генов, один из которых определяет развитие нормальных круглых глаз, а другой – полосковидных глаз. Скрещивается самка, имеющая полосковидные глаза, с круглоглазым самцом. Все потомство F1 имеет полосковидные глаза. Возвратное скрещивание самок из F1 с родителем привело к появлению потомства F2, в котором половина самок и половина самцов имело полосковидные глаза, а другая половина – круглые. Объясните характер наследования данного признака.
Потемнение зубов – доминантный признак, сцепленный с Х -хромосомой. У родителей, имеющих темные зубы, родилась дочь с темными и сын с белыми зубами. Какова вероятность рождения детей с белыми зубами в этой семье?
В развитых странах половина зарегистрированных случаев слепоты или слабовидения у детей вызывается генетическими заболеваниями, но, вероятно, это число занижено. Во многих развивающихся странах, где инвалидность детей по зрению встречается значительно чаще, генетические заболевания также являются важной группой состояний, часто вызывающих слепоту у детей.
«Генетическими» в этом контексте называются моногенные (менделевские) состояния. Многие проблемы диагностики и консультирования являются общими для всех заболеваний этой группы, что позволяет применять общий подход к ведению таких пациентов. Однако большая роль генетических факторов в развитии часто встречающейся патологии, например, была выявленная роль наследственных вариантов путей активации комплемента в патогенезе ВМД, и в наследовании нормальных количественных показателей (толщина роговицы, размер зрительного нерва), подтверждает наблюдение, что влияние молекулярных генетических факторов не ограничиваются менделевским наследованием.
Успехи в изучении наследственных заболеваний глаз являются одним из значительных достижений современной молекулярной генетики, от описания сцепления x1RP до идентификации первого гена adRP, кодирующего родопсин. Проект «Геном человека» ускорил изучение молекулярных механизмов генетических заболеваний. В настоящее время описано более 200 генных локусов и 150 генов, ответственных за моногенные заболевания сетчатки, 20 лет назад такое глубокое понимание патогенеза было недостижимо.
Геном человека распределен между 46 (23 парами, человек имеет диплоидный набор) физически различными хромосомами. Существует 22 пары аутосом, плюс две половые хромосомы: две X хромосомы у женщин, одна X и одна Y хромосома у мужчин. Хромосомы человека сильно отличаются друг от друга размером и формой, и гены, мутации которых вызывают моногенные болезни глаз, распределены между хромосомами без какой-либо закономерности.
Аутосомно-доминантные (autosomal dominant — AD) болезни вызываются мутациями генов хромосом 1-22. Больные имеют одну нормальную и одну мутантную копию гена (т.е. заболевание манифестирует в гетерозиготном состоянии). В большинстве семей с аутосомно-доминантными болезнями имеются несколько поколений мужчин и женщин, пораженных в одинаковой степени, отмечается наследование болезни по мужской линии.
Шансы больных передать мутантный ген каждому потомку составляют один к двум, независимо от пола. Риск заболевания потомка здорового пациента такой же, как и в общей популяции, при условии, что здоровый пациент точно не является носителем мутантного гена.
а) Экспрессивность. Члены одной семьи, пораженные моногенной болезнью, являются носителями одной генетической аномалии. Однако проявления патологического состояния могут варьировать в широких пределах. В этом случае говорят, что заболевание или, правильнее, мутантный аллель, характеризуется вариабельной экспрессивностью.
В качестве примеров можно привести синдром Марфана, неврофиброматоз I типа и окулокутанный альбинизм, при которых глазные и экстраокулярные проявления заболевания у носителей мутации варьируют в широких пределах. Фенотипически тяжесть заболевания носителя очень мало или вообще не влияет на прогноз заболевания у его сиблинга или потомка. Это ведет к неопределенности трактовки результатов консультативного или пренатального генетического обследования. Поэтому при аутосомно-доминантном наследовании важно обследовать родителей больного ребенка с целью выявления у них легких проявлений заболевания и прогнозирования 50% риска заболевания будущих потомков.
б) Пенетрантность. При некоторых аутосомно-доминантных болезнях вероятность развития клинической картины у носителей гена ниже 100% (т.е. мутация характеризуется сниженной пенетрантностью). Следовательно, при многих заболеваниях (например, различные формы аутосомно-доминантного пигментного ретинита (AD retinitis pigmentosa — adRP) колобома или врожденная катаракта), у носителей гена признаки заболевания могут отсутствовать, но риск развития заболевания у их потомков такой же, как и у потомков больных носителей.
Это еще одна причина для обследования родителей детей, например, с колобомой или дисгенезом переднего сегмента. Доступность генетических тестов способствует правильной оценке рисков.
в) Вновь возникшие мутации. Доминантные болезни могут также возникать de novo. В этом случае семейный анамнез не отягощен, заболевание возникает вследствие копирования дефекта ДНК одного из родителей. Это часто наблюдается при анирдии или ретинобластоме. В таких случаях риск развития заболевания у будущих сиблингов гораздо ниже 50%. Эта цифра не равна нулю из-за вероятности гонадного мозаицизма (т.е. у одного родителя мутация имеется в какой-то части сперматозоидов или яйцеклеток).
Трудно определить точную природу мутации, возникшей de novo — в случаях спорадической аниридии делеция может захватить и другие соседние гены. Это наблюдается при синдроме WAGR (аббревиатура от Wilms tumor, Aniridia, Genitourinary defect, mental Retardation — опухоль Вильмса, аниридия, дефект мочеполовой системы и умственная отсталость), когда делеция вызывает развитие опухоли Вильмса, аниридию, аномалии развития мочеполовой системы и умственную отсталость. Такое явление называется синдромом соседних генов.
Поэтому пациенту со спорадической аниридией необходимо или выполнять скрининговое ультразвуковое исследование почек, или подтвердить молекулярными методами, что ген опухоли Вильмса WT1 не изменен в результате новой мутации.
При возникновении новой аутосомно-доминантной мутации риск передачи ее потомкам пораженного пациента составляет 50%. Примеры таких состояний включают в себя редкие формы врожденного амавроза Лебера (вызываемые мутациями гена CRX) и ретинобластомы (вызываемой метациями в гене RB1). Поскольку мутация RB1 может характеризоваться сниженной пенетрантностью, наличие здоровых родителей может означать, что больной ребенок является носителем мутации, возникшей de novo, или что один из родителей является носителем мутации со сниженной пенетрантностью. Генетическое обследование поможет выявить членов семьи — носителей вызывающих заболевание генов и составить прогноз.
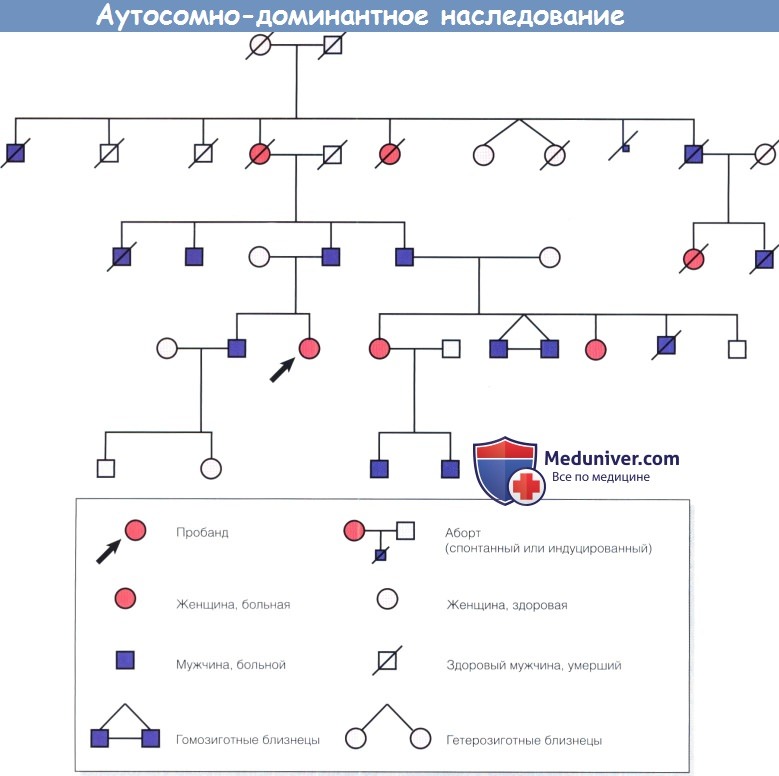
Родословное древо, иллюстрирующее аутосомно-доминантное наследование.
Редактор: Искандер Милевски. Дата обновления публикации: 18.3.2021
Хромосомная теория наследственности
Концепция данной теории заключается в том, что передача наследственной информации в ряду поколений осуществляется путем передачи хромосом, в которых в определенной линейной последовательности расположены гены.
Данная теория была сформулирована в начале XX века. Значительный вклад в ее развитие внес американский генетик Томас Морган.
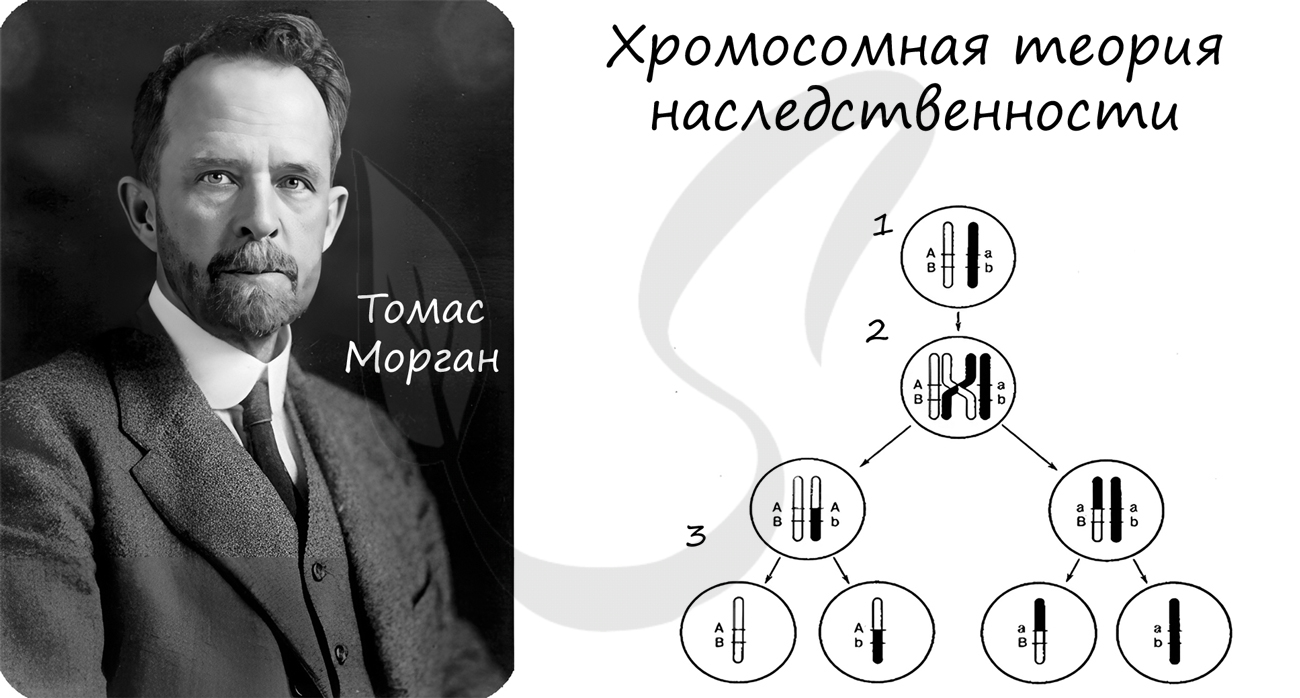
Рекомендую осознать и запомнить следующие положения хромосомной теории:
- Гены расположены в хромосомах в линейном порядке
- Каждый ген занимает в хромосоме определенное место - локус
- Гены, расположенные в одной хромосоме, образуют группу сцепления
- Сцепление генов может нарушаться в результате кроссинговера
- Частота кроссинговера между генами прямо пропорциональна расстоянию между ними
- Расстояние между генами измеряется в морганидах (1 морганида - 1% кроссинговера)
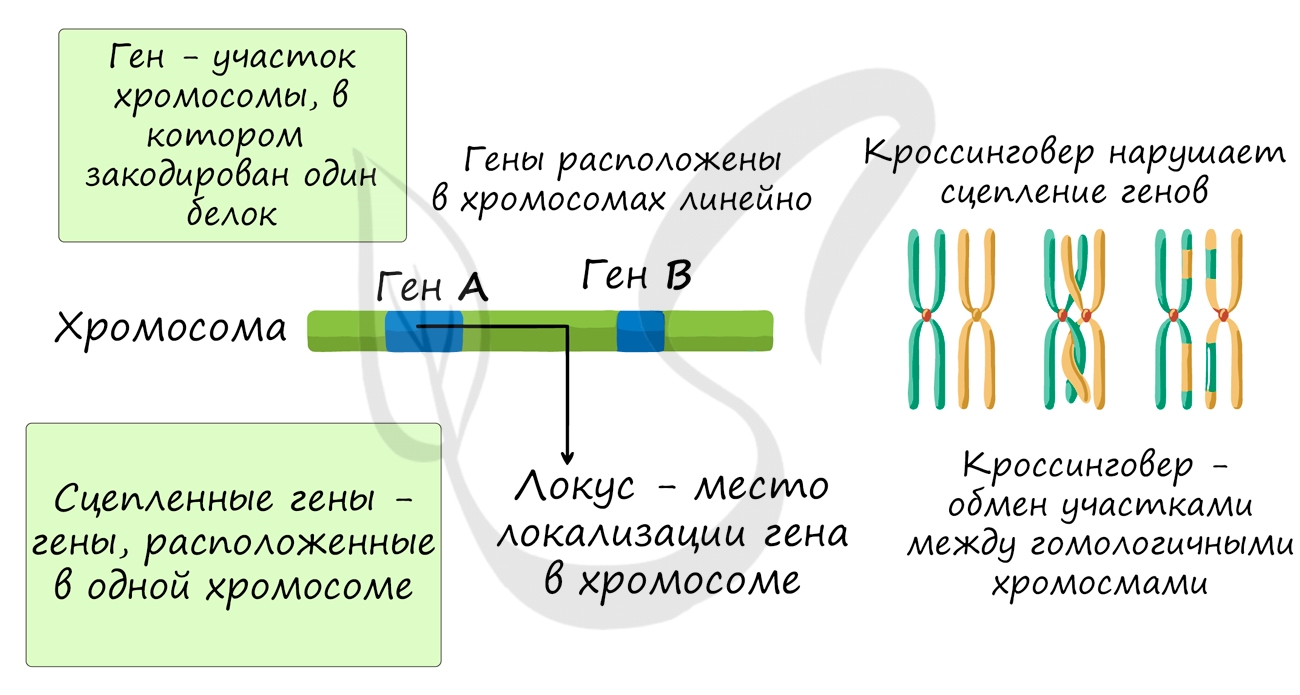
Группы сцепления
В предыдущей статье были раскрыты суть и применение в задачах III закона Менделя, закона независимого наследования, в основе которого лежат гены, расположенные в разных хромосомах. Но что если гены лежат в одной хромосоме? Такие гены образуют группу сцепления, в этом случае говорят о сцепленном наследовании.
Группа сцепления - совокупность всех генов, расположенных в одной хромосоме, вследствие чего они наследуются совместно. Число групп сцепления равно гаплоидному набору хромосом: у женщины 23 группы сцепления (23 пара - половые хромосомы XX), а у мужчины - 24 группы сцепления (X и Y представляют собой две отдельные группы).
Сцепление генов
Томас Морган в своих экспериментах изучал наследование признаков плодовых мушек дрозофил: серый (A) - черный (a) цвет тела, длинные (B) - зачаточные (b) крылья. В первом эксперименте Морган скрестил чистые линии плодовых мушек: серых с длинными крыльями (AABB) и черных с зачаточными (aabb).
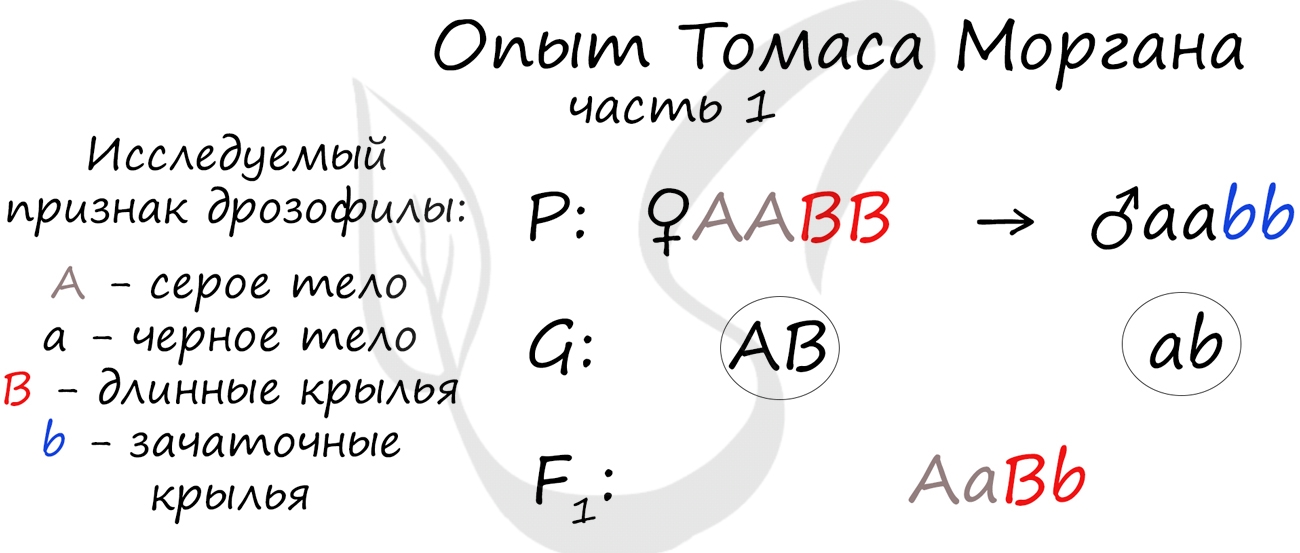
Только что вы видели первый закон Менделя (единообразия) в действии, правда, в несколько ином варианте - при дигибридном скрещивании. Но суть та же: в первом поколении все особи получаются единообразны по исследуемому признаку, с генотипом AaBb - с серым телом и длинными крыльями.
Далее Морган применил анализирующее скрещивание. Полученную в первом поколении дигетерозиготу (AaBb) он скрестил с черной особью с зачаточными крыльями (aabb). Результат весьма удивил Моргана и его коллег: помимо потомства с ожидаемыми фенотипами (серое тело + длинные крылья, черное тело + зачаточные крылья) были получены особи со смешанными признаками.

Потомство со смешанными признаками подразумевает под собой особи Aabb (серое тело + зачаточные крылья) и aaBb (черные тело + длинные крылья). Но откуда они могли взяться, если гены A и B находятся в одной хромосоме? Значит, образовались еще какие-то дополнительные гаметы, помимо AB и ab?
Объясняя полученные в потомстве фенотипы, которые содержали смешанные признаки, Томас Морган пришел к выводу, что между гомологичными хромосомами произошел кроссинговер, в результате которого образовались гаметы Ab, aB - кроссоверные гаметы.
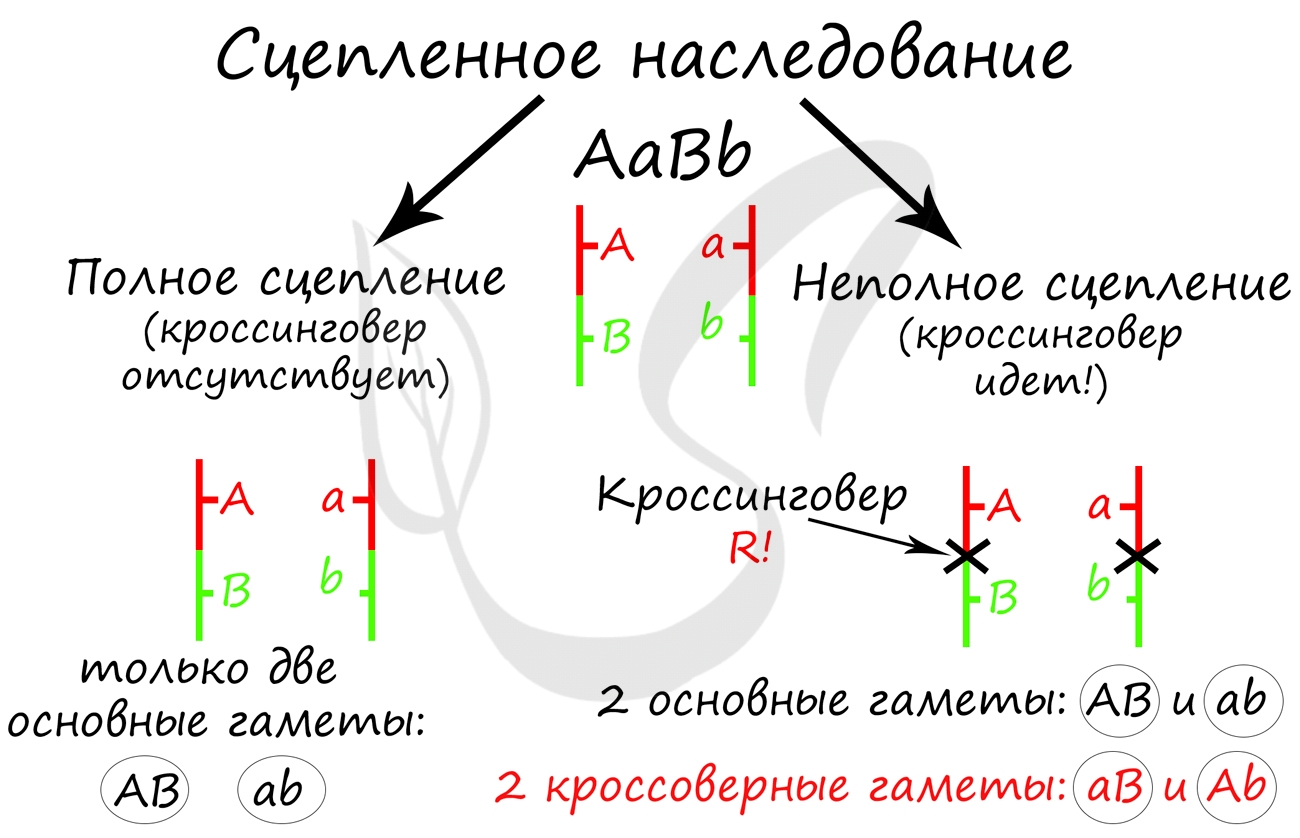
Очевидно, что в данном случае расстояние между генами A и B было 17 морганид, так как каждой кроссоверной гаметы (соответственно и особей) образовалось по 8.5%. Не забывайте, что процент кроссинговера равен расстоянию между генами. Поскольку расстояние было 17 морганид = 17%, то на каждую из кроссоверных гамет приходится половина - 8.5%
Пример решения генетической задачи №1
"Катаракта и полидактилия у человека обусловлены доминантными аутосомными генами, расположенными в одной хромосоме. Гены полностью сцеплены. Какова вероятность родить здорового ребенка в семье, где муж нормален, жена гетерозиготна по обоим признакам, мать жены также страдала обеими аномалиями, а отец был нормален".
Очень важно обратить внимание на то, что "гены полностью сцеплены" - это говорит об отсутствии кроссинговера, и то, что мы заметили это, обеспечивает верное решение задачи.

Самое главное, что вам следует усвоить: поскольку гены полностью сцеплены (кроссинговер отсутствует), женщина с генотипом AaBb может образовать только два типа гамет - AB, ab. Кроссоверные гаметы (Ab, aB) не образуются. Всего возможных генотипов потомков получается два, из которых здоров только один - aabb. Шанс родить здорового ребенка в такой семье ½ (50%).
Пример решения генетической задачи №2
"Гены доминантных признаков катаракты и эллиптоцитоза локализованы в 1-й аутосоме. Гены неполностью сцеплены. Женщина, болеющая катарактой и эллиптоцитозом, отец которой был здоров, выходит замуж за здорового мужчину. Определите возможные фенотипы потомства и вероятность рождения больного обеими аномалиями ребенка в этой семье".
Ключевые слова в тексте этой задачи, на которые следует обратить внимание: "гены неполностью сцеплены". Это означает, что между ними происходит кроссинговер.
Генотип женщины остается неясен из текста задачи. Раз она больна, то он может быть: AaBb, AABB, AABb, AaBB. Однако в тексте дано то, что развеет сомнения: "отец которой был здоров". Если ее отец был здоров, то его генотип был aabb, значит он передал дочери гамету ab. Теперь становится очевидно, что генотип дочери AaBb - она дигетерозиготна.

В данном случае между генами A и B произошел кроссинговер, их сцепление нарушилось. В результате образовались кроссоверные гаметы Ab, aB - которые привели к образованию особей с со смешанными признаками (Aabb, aaBb). Вероятность рождения в этой семье ребенка, больного обеими аномалиями, составляет ¼ (25%).
Наследование, сцепленное с полом
Половые хромосомы X и Y определяют пол человека. Генотип XX характерен для женщин, а XY - для мужчин. Мужская Y-хромосома не содержит аллелей многих генов, которые есть в X-хромосоме, вследствие этого наследственными заболеваниями, сцепленными с полом, чаще болеют мужчины.
Природа, несомненно, бережет женских особей. Женщины имеют две гомологичные хромосомы XX, и если ген наследственного заболевания попал в одну из X-хромосом, то чаще всего в другой X-хромосоме окажется "здоровый" ген, доминантный, которой подавит действие рецессивного гена. С генетической точки зрения, женщина будет носительницей заболевания, может его передать по поколению, но сама болеть не будет.
У мужчин если ген заболевания оказался в X-хромосоме, то не проявиться он не может. Именно по этой причине мужчины чаще страдают дальтонизмом, гемофилией и т.д.
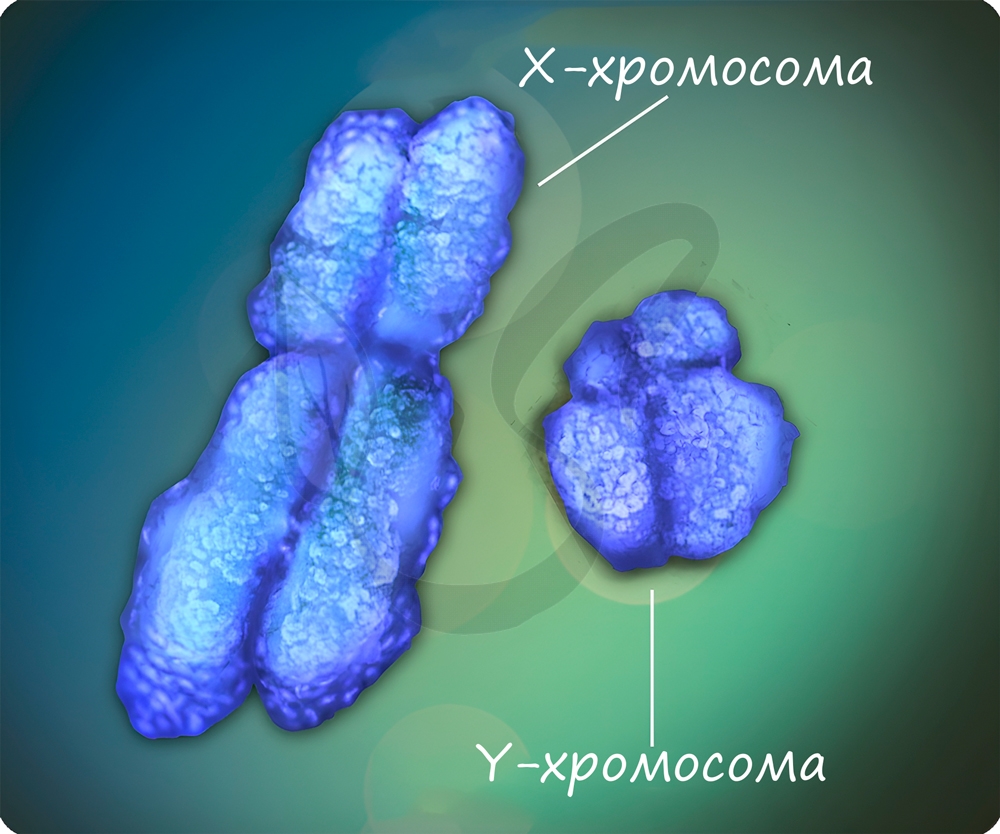
Не у всех организмов особь мужского пола характеризуется набором хромосом XY, а женского - XX. У пресмыкающихся, птиц, бабочек женские особи имеют гетерогаметный пол- XY, а мужские - XX. То же самое относится к домашним курам: петух - XX, курица - XY.
Решим несколько задач по теме наследования, сцепленного с полом. Речь в них будет идти о сцепленных с полом признаками - признаками, гены которых лежат не в аутосомах, а в гетеросомах (половых хромосомах).
Пример решения генетической задачи №3
"Рецессивный ген дальтонизма располагается в X-хромосоме. Женщина с нормальным зрением (отец был дальтоник) выходит замуж за мужчину с нормальным зрением, отец которого был дальтоником. Определите возможные фенотипы потомства".
Подробности о родословной важны и помогают заполнить белые пятна. Если отец женщины был дальтоником (X d Y), то очевидно, что он передал ей хромосому X d , так как от отца дочери всегда передается X-хромосома. Значит женщина гетерозиготна по данному признаку, а у мужчины возможен лишь один вариант здорового генотипа - X D Y. То, что его отец был дальтоником несущественно, ведь отец всегда передает сыну Y-хромосому.

Возможные фенотипы потомства:
- X D X D , X D X d - фенотипически здоровые девочки
- X D Y - здоровый мальчик
- X d Y - мальчик, который болен дальтонизмом
Пример решения генетической задачи №4
"Гипоплазия зубной эмали наследуется как сцепленный с X-хромосомой доминантный признак, шестипалость - как аутосомно-доминантный. В семье, где мать шестипалая, а у отца гипоплазия, родился пятипалый здоровый мальчик. Напишите генотипы всех членов семьи по данным признакам. Возможно ли у них рождение ребенка с двумя аномалиями одновременно?"
Ответ на вопрос: "Каковы генотипы матери и отца?" - лежат в потомстве. Пятипалый здоровый мальчик имеет генотип aaX b Y. Чтобы сформировался такой генотип, от матери должна прийти гамета aX b , а от отца - aY. Выходит, что единственно возможный генотип матери - AaX b X b , а генотип отца - aaX B Y.

Рождение ребенка с двумя аномалиями возможно - AaX B X b , вероятность такого события ¼ (25%).
Пример решения генетической задачи №5
"Рецессивные гены, кодирующие признаки дальтонизма и гемофилии, сцеплены с X-хромосомой. Мужчина с нормальным цветовым зрением и гемофилией женится на здоровой женщине, отец которой был дальтоником, но не гемофиликом. Известно, что мать женщины была гомозиготна по исследуемым признакам. Какое потомство получится от брака их дочери со здоровым мужчиной?"
Генотип мужчины вопросов не вызывает, так как единственный возможный вариант - X hD Y. Генотип женщины дает возможность узнать ее отец (X Hd Y), который передал ей гамету X Hd (отец всегда передает дочке X хромосому, а сыну - Y), следовательно, ее генотип - X HD X Hd
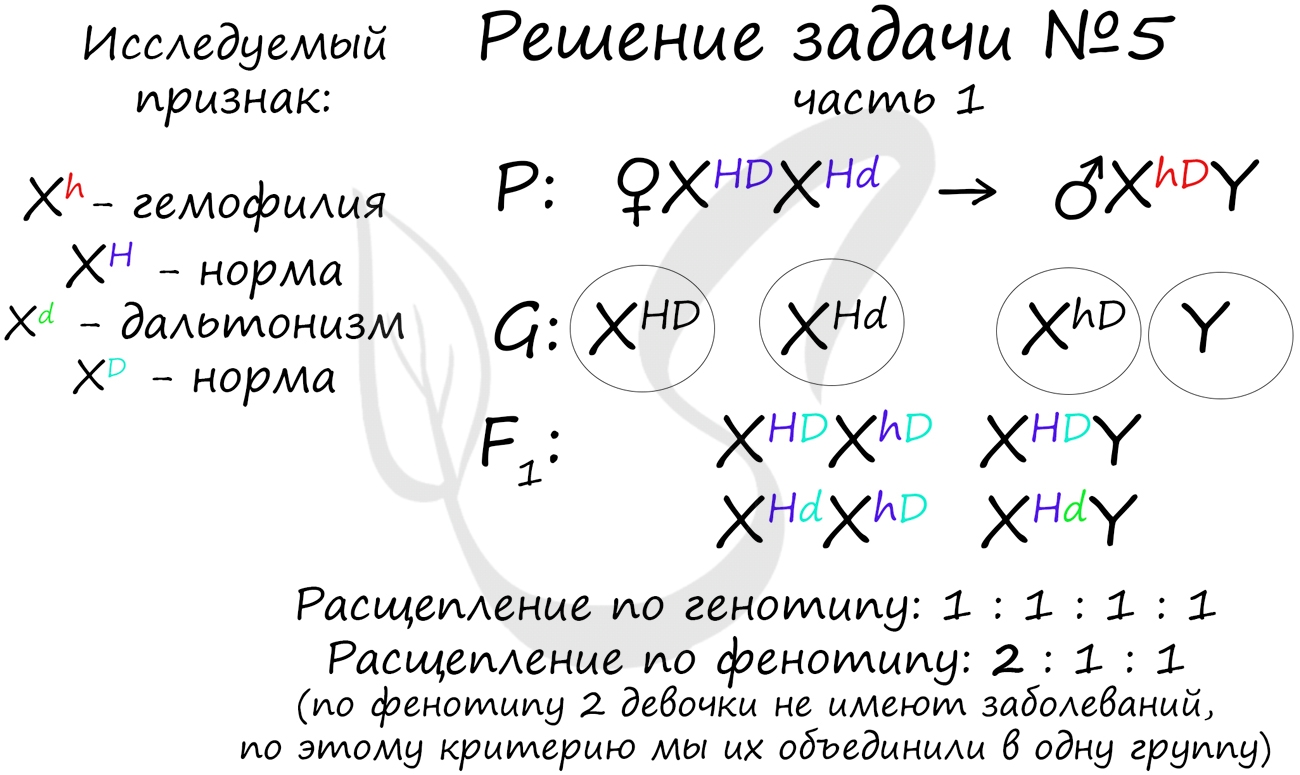
Как оказалось, возможны два варианта генотипа дочери: X HD X hD , X Hd X hD . Генотип здорового мужчины X HD Y. Следуя логике задачи, мы рассмотрим два возможных варианта брака.
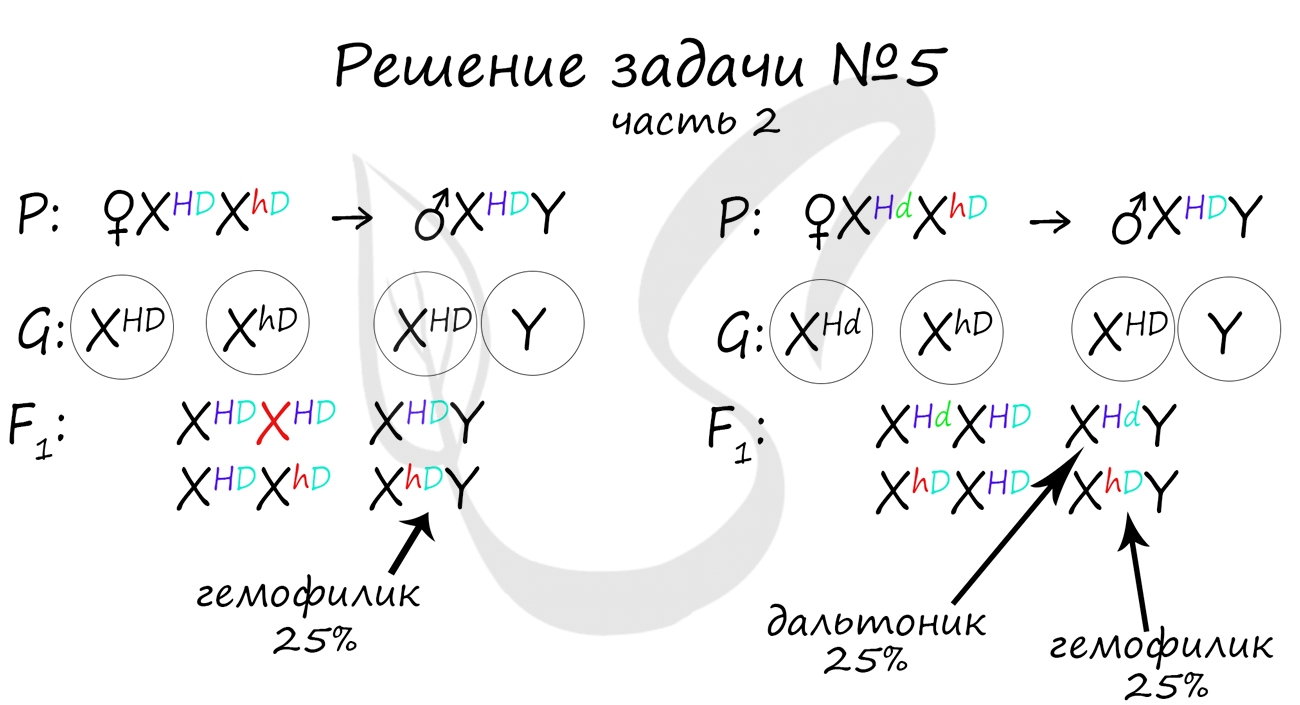
Не забывайте, что на экзамене схема задачи не является ответом. Ответ начинается только после того, как вы напишите слово "Ответ: . ". В ответе должны быть указаны все фенотипы потомства, их описание, что возможно покажется рутинными при большом числе потомков, но весьма приятным, если вы верно решили задачу и получили за нее заслуженные баллы :)
© Беллевич Юрий Сергеевич 2018-2021
Данная статья написана Беллевичем Юрием Сергеевичем и является его интеллектуальной собственностью. Копирование, распространение (в том числе путем копирования на другие сайты и ресурсы в Интернете) или любое иное использование информации и объектов без предварительного согласия правообладателя преследуется по закону. Для получения материалов статьи и разрешения их использования, обратитесь, пожалуйста, к Беллевичу Юрию.
Весь контент iLive проверяется медицинскими экспертами, чтобы обеспечить максимально возможную точность и соответствие фактам.
У нас есть строгие правила по выбору источников информации и мы ссылаемся только на авторитетные сайты, академические исследовательские институты и, по возможности, доказанные медицинские исследования. Обратите внимание, что цифры в скобках ([1], [2] и т. д.) являются интерактивными ссылками на такие исследования.
Если вы считаете, что какой-либо из наших материалов является неточным, устаревшим или иным образом сомнительным, выберите его и нажмите Ctrl + Enter.
- Код по МКБ-10
- Эпидемиология
- Причины
- Факторы риска
- Патогенез
- Симптомы
- Формы
- Осложнения и последствия
- Диагностика
- Дифференциальная диагностика
- Лечение
- К кому обратиться?
- Профилактика
- Прогноз
Относительно редкое заболевание – эктодермальная дисплазия – являет собой генетическое нарушение, сопровождающееся расстройством функциональности и структуры производных элементов наружного слоя кожи. Обычно поражаются волосы, ногтевые пластины, зубы, железистая система (слизистая, потовая и сальная). Болезнь сложная, может протекать в нескольких формах. Лечение преимущественно корректирующее, симптоматическое: к сожалению, о полном выздоровлении речь не идет. [1]
Код по МКБ-10
Эпидемиология
Вариациями эктодермальной дисплазии являются синдром Криста-Сименса-Турена, а также синдромы Клоустона, Рэппа-Ходжкина и ЕЕС. Патология впервые была описана в середине XIX века доктором Туреном. В 1913 и 1929 годах это описание было дополнено соответственно врачом-стоматологом Кристом и дерматологом Сименсом, в 1968 году – Рэппом и Ходжкиным, в 1970 году – Рудигером.
В медицинской литературе заболевание чаще встречается именно под названием эктодермальной дисплазии и соответствует международной кодировке Q82.4 (МКБ-10).
На сегодняшний день точную частоту заболеваемости врачи озвучить не могут. Однако считается, что синдром встречается примерно в одном случае из 5-10 тысяч. [2]
Точно известна этиологическая гетерогенность заболевания с тремя генетическими типами наследственной передачи: аутосомно-рецессивный, доминантный и Х-сцепленный рецессивный (последний – наиболее распространенный). [3]
В данный момент определены три гена в различных хромосомах, позволяющих выявить данное заболевание при помощи генетико-молекулярных методик. Численность возможных мутаций – более шестидесяти.
Эктодермальной дисплазией болеют чаще мальчики, что связано со сцепленным с полом наследованием. Девочки зачастую имеют легкую форму патологии, либо бессимптомную.
Патология регистрируется в разных странах мира у представителей любой расы. Может спорадически появляться у клинически здоровых пар, либо проявляться в семейной форме (особенно часто – если родители имеют близкородственные связи).
Причины эктодермальной дисплазии
Единственная причина развития эктодермальной дисплазии заключена в мутации определенного наследственного генного фактора. В частности, наиболее распространено нарушение со стороны гена EDA, локализованного на Х-хромосоме. Этот ген ответственен за кодировку белкового вещества эктодисплазин-А, неправильная структура которого влечет за собой нарушение формирования элементов эктодермы. В настоящее время точные характеристики белкового вещества и механизм развития мутационных нарушений не выяснены.
Заболевание, сцепленное с Х-хромосомой, имеет свои особенности: проблема обнаруживается чаще у мужчин, но и женщины способны выступать не просто носителями, но и иметь отдельные признаки синдрома, хоть и в нетяжелой степени. К примеру, у пациенток с эктодермальной дисплазией можно наблюдать избыточную сухость кожных покровов, морщинистость, истончение и сухость волос, зубную деформацию. Возможны проблемы с грудными железами и сосками. Подобные признаки указывают на возможность неполного доминирования генных мутаций EDA.
Среди других типов мутаций можно выделить изменения гена EDAR, который отвечает за кодировку рецептора к фактору опухолевого некроза. Этот ген локализуется на II хромосоме, наследование происходит аутосомно-рецессивно. В точности процесс развития патологии не выяснен.
Если речь идет о редких вариантах эктодермальной дисплазии, то они возникают под влиянием генных мутаций в TDARADD, ответственном за кодировку белка-рецептора к экзодисплазину-А, локализованному на I хромосоме. Патогенетические механизмы полностью не изучены. [4]
Факторы риска
Наиболее значимыми факторами риска, ведущими к рождению ребенка с эктодермальной дисплазией, являются дефекты:
- гена EDA, кодирующего эктодисплазин-А, картированного на хромосоме Xq12-q13.1;
- гена EDAR, кодирующего рецептор фактора опухолевого некроза, члена суперсемейства EDAR, картированного на хромосоме 2q11-q13;
- гена TDARADD, кодирующего эктодисплазин-А – рецептор-ассоциированный белок, картированного на хромосоме 1q42.2-q43.
Заподозрить наследственную предрасположенность к эктодермальной дисплазии можно при исследовании семейной истории.
Комплексная генно-молекулярная диагностика позволяет оценить генетический риск развития у ребенка данного синдрома.
Патогенез
Патогенетические особенности развития данного заболевания малоизученны. Известно, что эктодермальная дисплазия появляется в результате мутационных изменений в определенных генах. Причина наиболее частой формы патологии заключается в повреждении гена EDA, локализованного на Х-хромосоме. Этот ген отвечает за кодировку белкового агента с названием эктодисплазин-А. Патологические изменения в его структуре вызывают неправильное развитие эктодермальных производных. К сожалению, на сегодняшний день и функциональная сторона этого белкового агента, и патогенез изменений при мутировании гена EDA изучены недостаточно.
Основная отличительная черта эктодермальной дисплазии заключается в том, что клинические нарушения обнаруживаются не только у пациентов мужского пола, но и у женщин: носительство проявляется более легкими патологическими изменениями. В частности, отмечается сухость волос и кожных покровов, ранняя морщинистость, искривления и прочие нарушения со стороны зубов.
Помимо этого, причинами типичного синдрома Криста-Сименса-Турена выступают мутационные изменения в гене EDAR, ответственном за кодировку одного из рецепторов к фактору опухолевого некроза. Этот ген локализован на II хромосоме, наследование происходит аутосомно-рецессивно. Патогенетические особенности и в этом случае не изучены. [5]
Известна и более редкая разновидность ангидротического типа эктодермальной дисплазии, с аутосомно-доминантным способом наследования. Причиной выступают мутационные изменения гена TDARADD, кодирующего белковое вещество-рецептор к экзодисплазину-А и локализованного на I хромосоме. Скорее всего, патогенетические характеристики в данном случае идентичны с более распространенной разновидностью заболевания, сцепленной с полом.
К сведению: эктодерма представляет собой один из трех зародышевых листков (ещё два представлены мезодермой и энтодермой). Эктодерма – это наружный слой, который формируется на протяжении третьей недели эмбрионального развития и обусловливает образование кожных покровов и придатков (волосы, ногти), прямокишечного и ротового эпителия, зубной эмали, хрусталика и роговицы, потовыделительных желез. У людей, страдающих эктодермальной дисплазией, некоторые или все структуры эктодермы, либо отсутствуют, либо недостаточно функционируют.
Симптомы эктодермальной дисплазии
Клиническая картина при эктодермальной дисплазии определена целым рядом многочисленных нарушений, затрагивающих эктодерму и потовые железы. Поражаются также сальные и апокриновые железы, но эти дефекты менее выражены. Другие железистые системы – в частности, слезные, пищеварительные, носовые, бронхиальные – имеют признаки атрофии. Типичные признаки: атрофическое процессы, гипоплазия кожных покровов, гипоплазия грудных желез и сосков.
В области лица обнаруживаются морщины, истончение век, периорбитальное нарушение пигментации, папулы, экзематозные высыпания, ладонный гиперкератоз. Лобные бугры и надбровные дуги явно выдаются вперед, переносица сглажена, нос небольшой седловидный, носовые крылья гипопластичны, губы полные и выступающие, щеки – впалые.
Волосяной покров редкий, часто – с залысинами, отличается сухостью и светлыми оттенками.
Зубы имеют неправильную конфигурацию, часто – коническую заостренную форму. Некоторые зубы вовсе отсутствуют (клыки присутствуют всегда).
Ушные раковины также деформированы: обычно они маленькие, высоко посаженные, ушной завиток имеет неправильную форму.
Со стороны органов зрения может отмечаться помутнение хрусталика, близорукость, блефароконъюнктивит, снижение слезовыделения, жидкое стекловидное тело.
Некоторые пациенты страдают полным отсутствием слуха. Отмечается склонность к инфекционным заболеваниям, нарушениям терморегуляции.
Первые признаки
Первые проявления эктодермальной дисплазии зачастую обнаруживаются уже в периоде новорожденности. Однако это может произойти и позже, так как клиническая картина у маленьких детей не всегда носит выраженный характер и усугубляется с годами.
Базовыми симптомами, по которым можно заподозрить наличие патологии, часто становятся такие:
- отсталость роста на фоне относительно большой головы;
- сухость, тонкость волос, преимущественно «пушковые» волосы с замедленным ростом и малой пигментацией, укороченность и редкость ресниц и бровей, либо полное их отсутствие;
- ранняя алопеция, вплоть до полной потери волосяного покрова;
- типичный внешний вид по типу «стариковского лица», выступающая лобная область, надбровные дуги и бугры, расширенные скулы, западение переносицы, небольшой седлообразный нос и гипопластические крылья, впалые щеки, выдающиеся губы по типу «рыбьих», «тяжелый» подбородок, неправильная форма ушных раковин;
- запоздалое прорезывание зубов (от года до трех лет), нарушение привычной последовательности прорезывания, длительный период сохранения молочных зубов, отсутствие некоторых зубов;[6]
- конусовидная зубная конфигурация, заостренные режущие края, сглаженная поверхность моляров;
- нарушения зубного ряда и прикуса;
- недостаточно развитые слюнные железы, слабое слюновыделение, сухость во рту, сиплость;
- чрезмерная сухость кожи, ранняя морщинистость, что особенно заметно на лице;
- нарушения пигментации, неправильная работа сальных желез, папулезная сыпь;
- конъюнктивиты, светофобия;
- недоразвитые грудные железы, либо их отсутствие;
- недостаточно развитые слизистые железы в респираторной и пищеварительной системе, что обусловливает частые бронхиты, риниты, синуситы, желудочно-кишечные патологии;
- периодическое резкое повышение температуры, связанное с неправильной теплоотдачей из-за нарушения работы потовых желез;
- реже – умственная отсталость, олигофрения (чаще развитие интеллекта соответствует норме);
- нарушенная социальная адаптация и ориентация, скованность и замкнутость;
- речевые проблемы, связанные с неправильным зубным ростом и сухостью слизистых ротоносоглотки;
- нарушенное зрение;
- слабое или отсутствующее потовыделение.
Триада при ангидротической эктодермальной дисплазии
Ангидротический вариант эктодермальной дисплазии проявляется триадой базовых признаков:
- редкое оволосение по типу атрихоза или гипотрихоза; [7], [8]
- неправильная конфигурация зубов (часто – коническая, заостренная), либо недоразвитие и отсутствие зубов;
- нарушения потовыделения по типу гипо и ангидроза, что часто обусловлено отсутствием потовых желез как таковых.
Ввиду наличия ангидроза у больного отмечаются такие вспомогательные признаки, как высокотемпературная гиперчувствительность и регулярные рецидивы гипертермии, что представляет собой реальную опасность для жизни человека. Кожные покровы истончены, отличаются сухостью. Многие больные страдают хроническим блефароконъюнктивитом, «синдромом сухого глаза», астмоподобными состояниями. [9]
Формы
Разные совокупные проявления и их интенсивность определяют подразделение эктодермальной дисплазии на несколько видов, которые можно назвать самостоятельными формами патологии. Основными такими видами считаются: синдром Криста-Сименса-Турена, синдром Клоустона, синдром Рэппа-Ходжкина и синдром ЕЕС.
Синдром Криста-Сименса-Турена, или ангидротическая эктодермальная дисплазия характеризуется полной дисфункцией потовых желез, а также своеобразным фенотипом строения лицевой области: у ребенка обнаруживается выдающийся кпереди лоб, утонченные и негустые брови, редкие короткие ресницы, морщины. Типично периорбитальное нарушение пигментации, седлоподобная переносица, челюстная гипоплазия. Волосы бывают депигментированными, либо слабопигментированными.
Некоторые специалисты посчитали, что полномерный ангидроз у пациентов все же обнаруживается редко, и у большинства больных потовыделительная система слабо, но все-таки функционирует. Это мнение было учтено и привело к тому, что врачи со временем стали использовать более корректное название: гипогидротическая форма заболевания. Гипогидротическая эктодермальная дисплазия представляет собой генетическое расстройство формирования эктодермального слоя. Патология характеризуется нарушениями образования таких элементов эктодермы, как кожные и волосяные покровы, железы (потовые, сальные) и зубы. Болезнь состоит из трех подтипов, которые практически не отличаются симптоматически, так как основным клиническим признаком становится нарушенное потовыделение (преимущественно гипогидроз). Речь идет о непосредственно синдроме Криста-Сименса-Турена с Х-сцепленным типом наследования, а также аутосомно-рецессивной и аутосомно-доминантной эктодермальной дисплазии. Существует ещё несколько менее распространенных подтипов, сопровождающихся выраженным дефицитом иммунитета – так называемая врожденная ангидротическая эктодермальная дисплазия с иммунодефицитным состоянием.
Синдром Клоустона представляет собой гидротический тип эктодермальной дисплазии. Определяющими симптомами патологии становятся все те же поражения зубов, волос и потовыделительной системы, но в несколько меньшей степени. Гиподентия обнаруживается в области нижних резцов, вторых моляров и верхних клыков. Ногтевые поражения проявляются в виде гипоплазии, дистрофии, аплазии с паронихиями. Снижена численность потовыделительных желез, при неизменных сальных железах. Возможен гипотрихоз, облысение. Способ наследования аутосомный и аутосомно-доминантный.
Синдром Рэппа-Ходжкина иначе называют гипогидротической эктодермальной дисплазией, сопровождающейся расщелиной губы, альвеолярного отростка, мягкого и твердого неба. Отличительными проявлениями считаются такие: гипогидроз и гипотрихоз, патологические изменения ногтей, гиподентия или олигодентия в соединении с расщелиной верхней губы, альвеолярного отростка, мягкого и твердого неба. Общими симптомами становятся также впадение переносицы, сужение носа, микрогнатия верхней челюсти, небольшой рот, уменьшенные гениталии. Синдром наследуется по аутосомно-доминантному типу.
Синдром ЕЕС лишь недавно выделили в качестве самостоятельного заболевания, более известного как совокупный синдром эктродактилии, эктодермальной дисплазии с расщелиной неба и верхней губы. Отличительными симптомами являются дефекты стоп и кистей, расщелина верхней губы, иногда – расщелина языка. Эти признаки присутствуют на фоне нарушения потовыделения, гипотрихоза и алопеции, ногтевой гипоплазии, сухости и гипопигментации кожи, конъюнктивита, светофобии и пр. Типичны также зубные аномалии, позднее прорезывание, множественный кариес. На фоне физических дефектов умственное развитие обычно адекватное. Способ наследования – аутосомно-доминантный, но встречаются и рецессивно наследованные варианты.
Эктодермальная дисплазия у детей
Несмотря на то, что эктодермальная дисплазия является врожденным заболеванием, её не всегда удается диагностировать у новорожденного малыша: зачастую диагноз ставят лишь через несколько лет (часто – к 2-3 годам). Практикующие врачи отмечают необходимость ранней диагностики, так как от этого может зависеть не только дальнейший образ жизни, но иногда и непосредственно жизнь больного. Симптомы патологии многообразны, но не всегда заметны. При этом некоторые из них встречаются чаще, а другие – реже. [10] И родителей, и врачей должны насторожить такие признаки:
- гипоплазия потовыделительных желез с гипо или ангидрозом, терморегуляторные нарушения, частые эпизоды гипертермии, беспричинная лихорадка, регулярные перегревы;
- явления гипотрихоза, редкость, депигментация и тонкость волос, укорочение бровей и ресниц (или их отсутствие);
- устойчивое или преходящее облысение (полное или очаговое);
- позднее зубное прорезывание с нарушением его последовательности;
- недостаток количества зубов, нарушение их конфигурации (часто – конусообразная, шиповидная форма с заостренным краем), либо отсутствие зубов;
- нарушения прикуса, иногда – подвижность зубов, большие межзубные промежутки;
- низкое прикрепление верхнегубной уздечки, резкая выраженность щечных тяжей, мелкое ротовое преддверие;
- недостаточно развитый верхнечелюстной альвеолярный отросток;
- на рентгенограмме – укороченные зубные корни, расширенные периодонтальные щели, уплощенные мыщелковые нижнечелюстные отростки;
- гипоплазия слизистых желез во рту, как результат – недостаточное слюновыделение, сиплость;
- склонность к грибковым стоматитам, хейлитам;
- типичное «лицо старика» с выдающейся лобной областью, запавшей переносицей, небольшим седлообразным носом, впалыми щеками, полными нечеткими выпуклыми губами, неправильной формой ушных раковин;
- истонченная сухая морщинистая кожа, иногда – с папулезной сыпью;
- недостаточная функция слезных желез, частые воспалительные заболевания (кератиты, блефариты и пр.);
- дефекты губы и неба;
- ногтевые поражения, паронихии;
- дефекты стоп и/или кистей, гиперкератоз ладоней и стоп;
- недостаточное развитие грудных желез и сосков (иногда – их отсутствие);
- иммунодефицит, экзема;
- склонность к респираторным и пищеварительным заболеваниям, а также к носовым кровотечениям.
Разные сочетания признаков и их интенсивность определяют отдельные варианты течения эктодермальной дисплазии.
Читайте также: